Learning is extremely difficult for children with Angelman syndrome. Developmental milestones such as sitting up or speaking come late. As they get older, children with the syndrome have trouble walking and sleeping through the night, and suffer from seizures.
But the most prominent symptom of Angelman syndrome is impaired speech. Normally children learn up to three thousand words a year while they’re in school. But many children with Angelman learn only one or two words over their lifetimes; most never learn to speak at all. With a lot of instruction, people with the syndrome can learn to do simple tasks, but they never become completely independent, and require lifelong care.
Philpot suspected that the source of the learning deficits was the brain’s inability to process sensory experience. “Your sensory experiences leave a trace on the connections of your brain that can be recalled later on,” he says. “All of our experiences modify our brains in some way. Sometimes the modifications are simple and sometimes they are robust.”
Essentially, brains develop like paintings, colored over time by the sights, sounds, and smells that we experience. Each sensation is like a brush stroke, adding a new detail to the emerging picture. In the brain, these sensations stimulate formation of new connections between neurons, ultimately changing how the brain is wired. But with Angelman syndrome, the colors drip out of place. Instead of developing a clear portrait of the world, the brains of children with Angelman form a muddled collage.
Scientists have known for years that Angelman syndrome is caused by mutation of the UBE3A gene. UBE3A instructs the body to destroy unnecessary or damaged proteins. A mutation that deletes UBE3A leads to Angelman syndrome. But relatively few studies have examined how a lack of UBE3A could affect brain activity.
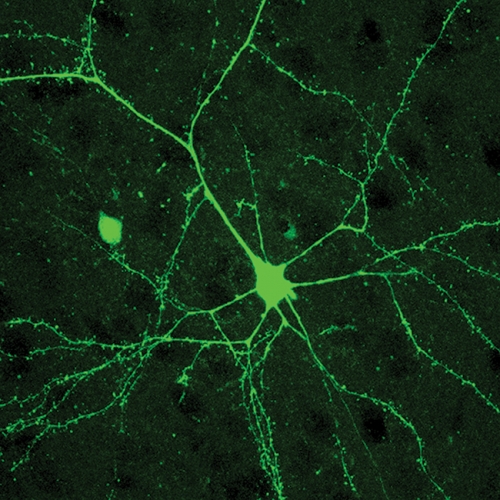

Philpot’s group began to study mice that lacked UBE3A and had learning deficits similar to those in human patients. Koji Yashiro, a graduate student in the lab, took electrical recordings of neurons using electrophysiology. Like an electrician who uses a meter to check the current running through wires, Yashiro used microscopic electrodes to measure the activity of neurons. Based on these recordings he was able to determine how well neurons were connecting to each other. He also measured the plasticity of the connections, which is the ability of the connections to change in response to experience. Not surprisingly, the neurons in the mice with Angelman-like symptoms had severely impaired plasticity and had fewer connections than neurons in the normal mice did.
Most of Yashiro’s recordings were performed in the visual cortex, a part of the brain where neurons process visual signals from the eyes. But how can the visual system reflect the severe learning deficits in Angelman syndrome? “By studying sensory experiences and how they modify visual areas of the brain, we can determine how they might affect learning in other areas,” Philpot says.
The visual system is so well studied that scientists have developed a map of the visual cortex. They know the exact type of visual cues that activate different parts of the cortex, and so they can study how the brain is affected by sensory input. Hearing, touching, tasting, and smelling are difficult to manipulate in live animals, but vision can be changed simply by putting animals in a dark room.
The scientists wanted to understand how a complete lack of visual experience would affect the study mice’s neuron activity, and so they raised the animals in the dark. Yashiro expected the mice to show a little improvement once their vision was impaired, but what he found was startling: the neuronal connections in Angelman-syndrome mice regained their plasticity.
“It looked like what was causing the defect was sensory experience,” he says. “This finding is a catch-22. In order to keep the mice normal you have to deprive them of sensory experience, but without sensory experience their brains won’t develop properly. It would be tough to develop a treatment based on this finding.”
But Philpot believes that there could be other treatment approaches. For example, patients with Angelman may be able to learn tasks better if they are in a limited environment with minimal distractions. Another option, since Yashiro has shown that the study mice are capable of normal brain plasticity, is to develop a medication that could prevent damage from sensory experience. But researchers first have to determine which molecular changes are causing the damage.
“We don’t know too much about what UBE3A is doing,” Philpot says. UBE3A marks a lot of proteins for destruction. So the undestroyed proteins have to be the source of the problem. The question is, which ones? Philpot’s group is working on a project that will help them identify the harmful proteins. They are also participating in a preliminary drug-discovery project; they hope that certain compounds may be able to reverse the defects in the study mice.
UBE3A has connections to another devastating neurological disorder. “Too little UBE3A and you get Angelman syndrome,” Philpot says. “Too much of it and you get autism.” According to Philpot, 5 percent of autism cases have been linked to excess amounts of UBE3A. Scientists aren’t sure how a high amount of UBE3A leads to autism. But the idea that Angelman syndrome and autism are on the same spectrum means that researchers may be able to develop UBE3A-based treatments to target both disorders.
New treatments will take several years to develop, but Philpot believes that there is more hope for Angelman syndrome patients now than ever before. The dominant idea in Angelman syndrome research has been that without UBE3A, the brain would never be able to function normally. Now that Philpot’s study has shown that the brains of mice with Angelman-like symptoms are capable of normal plasticity, there may be more interest in finding a way to restore brain activity in humans.
A smile spreads across Philpot’s face as he thinks of the possibilities. “Over my seventeen years in neuroscience,” he says, “this is the most striking finding I have ever observed.”
Meagen Voss received a master’s degree in neurobiology in spring 2010.
Ben Philpot is an assistant professor in the UNC Neuroscience Center and in the Department of Cell and Molecular Physiology in the School of Medicine. Koji Yashiro, the primary author of this work, earned a doctorate from the School of Medicine in 2008. Their study was published in the journal Nature Neuroscience. Funding came from the National Institutes of Health, the Angelman Syndrome Foundation, and the Simons Foundation.