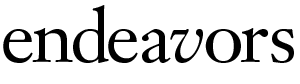


“It’s not cancer.”
Those are the words Dianne Shaw heard as she was being wheeled out of the operating room. “I thought, ‘Good, now I can get back to my life,’” she says. “What I didn’t hear the doctor say was ‘But we don’t know what’s causing the spot on her lung.’”
A few weeks later, Shaw was started on therapy for a possible viral infection. Then came a day when she couldn’t get out of bed. “All I could eat was cherry Jell-O,” she says. At the hospital, doctors said her lungs were bleeding massively. Two days later, Shaw was finally diagnosed with Wegener’s granulomatosis. It’s an autoimmune disease, which means that, somewhere inside her, a cell has signaled Shaw’s immune system to attack her own body. In Wegener’s, antibodies inflame the walls of the small blood vessels. The inflammation restricts blood flow to various organs, damaging them. If Wegener’s isn’t diagnosed and treated, it can be fatal.
Shaw was diagnosed nine years ago. To control her symptoms, she has taken anticancer drugs, huge doses of prednisone, and other immune-system suppressants. The drugs dampen the inflammation in her body. But each drug works for only a while. “She’s been on everything,” says Shaw’s doctor, Ron Falk, Chief of the Division of Nephrology and Hypertension in the School of Medicine.
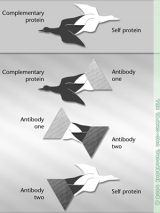
The immune-system suppression leaves Shaw vulnerable to infections, and some of the drugs put her at increased risk for glaucoma, cataracts, osteoporosis, diabetes, and cancer. She has had four sinus surgeries because of massive infections and scarring. After three ear surgeries, she wears hearing aids in both ears because of scarring damage. She has undergone twenty-one surgeries to keep her airway open. At times, her trachea narrowed to the size of a drinking straw. “But I’m lucky,” she says. “I have the best care available. And the disease hasn’t hit my kidneys.” Many people with Wegener’s eventually need kidney transplants.
Broad immune-system suppressants are Shaw’s only choices for treatment because the cause of autoimmune diseases remains mysterious. Scientists know that Wegener’s and some other autoimmune diseases such as lupus begin with an antibody — a protein that normally fights disease. For some reason the body produces an antibody that binds to a normal protein — a so-called self protein. This traitorous antibody is called an autoantibody. If scientists could find out exactly where on a particular self protein an autoantibody binds, they’d be on their way to developing more specific treatments.
When Gloria Preston, now a research associate professor, joined Falk’s team in 1998, her predecessors had been looking for that binding spot. Researchers had taken blood samples from patients with Wegener’s disease and combined the samples with different pieces of a normal protein, PR3, which is a known target of autoantibodies. The researchers were trying to find out which piece of the protein the patients’ autoantibodies would bind to.
“If one could identify that piece, and say yes, the autoantibody binds there, then one could develop a therapy to block that interaction,” says Will Pendergraft, a postdoctoral fellow in the nephrology division.
Out of those first experiments came an unexpected result — something that many scientists might have ignored. It led Falk’s team to a new theory about how autoimmunity works.
To make pieces of PR3 protein, the researchers put pieces of PR3 DNA into a bacterial expression system — a way to quickly produce pieces of the protein. The researchers grew thousands of colonies of bacteria. To each colony they added a different piece of DNA, so each colony would produce a different piece of PR3 protein. Then they added blood serum from Wegener’s patients. When they saw that serum reacted with a particular colony, they’d sequence the protein expressed there to find out which piece of PR3 the patients’ antibodies had bound to.
But results showed that some antibodies were acting strangely. They were binding to some pieces of protein that had a backwards sequence — opposite from the sequence of PR3. These pieces were created just as a by-product of the experiment. They were parts of PR3’s antisense protein — a complementary protein that is coded for by the noncoding strand of DNA.
That was unusual because scientists have long thought that the noncoding strand of DNA isn’t used in humans. In fact, for many years scientists assumed that the noncoding strand didn’t have much function at all, except to act as a template when DNA copies itself.
In the past few years that thinking has changed. It had to. Scientists have found that the portion of DNA that codes for proteins makes up less than 2 percent of the total DNA in the human body. The other 98 percent has to be good for something.
But the purpose of the rest of our DNA, including that noncoding strand, is pretty mysterious. Scientists know that in bacteria, both strands of DNA are used to produce proteins, Preston says. Recent studies have shown that even humans make antisense RNA transcripts from noncoding DNA.
But the times haven’t changed that much. “No one believes any of the human antisense transcripts are ever translated into proteins,” Preston says. So in the Carolina studies, antibodies in the patients’ blood seemed to be binding to a type of protein that most scientists don’t believe is likely to exist.
That was the story when Preston came to Carolina. Falk showed her the data and asked her to check it out. “Many scientists might have just thrown out these results because they didn’t fit with the dogma of biochemistry,” Pendergraft says. If the results were correct, Preston says, “patients could be making antisense proteins as an initiating event in autoimmunity. That’s an unheard-of idea.”
But the aberration kept showing up so many times that Falk couldn’t ignore it. He read a paper called “Making sense from antisense,” by Alex Tropsha, associate professor in the School of Pharmacy, published in the Journal of Molecular Recognition. Tropsha’s paper reviewed studies showing that some proteins made by the coding strand and those made by the noncoding strand have been known to interact. These proteins are opposites in many ways, but they have complementary structures — they fit like a hand and glove. So these mirror-image proteins had been shown to bind to each other. This relationship wasn’t well known. But it had been seen.
When Falk showed Preston that paper, she was intrigued. “I had never heard of it. It’s not something you read in biochemistry textbooks,” she says. The more traditional idea is that protein interactions are driven by the sequences of amino acids (the building blocks that make up proteins).
“So we set out to put these earlier observations to the test,” she says. She supervised the experiments, and Pendergraft, then a Ph.D. student, did much of the hands-on work. He took on the project as one possibility for his dissertation work. Not that he expected a whole lot. It was a long shot.
First, the scientists wanted to make sure that they were really seeing what they thought they were seeing. Remember that, in the previous experiments, to produce the pieces of PR3 protein, the researchers had put PR3 DNA into bacterial cells. This is a quick way to do an experiment because bacteria grow rapidly. “You can get an answer to a scientific question overnight,” Pendergraft says. But maybe, since humans are exposed to bacteria all the time, the patient blood was really reacting to some nonspecific bacterial protein, not to the antisense protein. “We couldn’t be completely sure what the human antibodies were binding to,” Pendergraft says.
So they did new, “cleaner” experiments. Instead of using bacterial cells to produce the protein, the researchers used human cells. This took longer. But they could be sure that any binding was not an artifact of the way they were producing the proteins.
In those experiments, they again saw binding of human antibodies to the antisense protein, called cPR3 (c for complementary). “And not just one or two times,” Preston says. “The results were very reproducible.” Maybe there really was something going on here.
To be sure, the researchers wanted to do many more tests. But first they needed a batch of purified cPR3. Pendergraft couldn’t just call a medical-supply company and order some. He had to make it himself. In the end, it took him two years to make enough of it to use in their experiments.
Why so long? This protein turned out to be hard to manipulate. Preston explains that, though sense proteins and antisense proteins are mirror images structurally, in most other ways they are exact opposites. So if the sense protein is easily dissolved in water, for instance, then its antisense protein is not.
By the fall of 2001, Pendergraft had made enough of the purified cPR3. He and Preston performed additional experiments, including an ELISA test, an experiment that was more controlled than the previous ones. For the ELISA experiments, the researchers used blood samples from four groups — one group of patients with Wegener’s and three other groups used as controls. They combined the blood samples with just one piece of the antisense protein.
In the three control groups, none of the patient blood reacted. But in the Wegener’s group, seven out of thirty-four patients had antibodies in their blood that reacted with the piece of cPR3. “We were ecstatic because we knew the implications,” Pendergraft says. Preston says, “The fact that we found these antibodies in patients at all was amazing.”
After repeating their findings, the researchers devised a new theory about how some autoimmune diseases work. The trigger for autoimmunity is not the well-known self protein but its mirror image — the antisense protein.
Somehow the antisense protein is made inside the body, or maybe it enters from outside. The body tries to get rid of the antisense protein by making an antibody that will bind to it. This is where it gets complicated. In reaction to that first antibody, the immune system creates a second one — an anti-antibody.
This creation of antibodies and anti-antibodies, and even a third generation, anti-anti-antibodies, is how the immune system keeps itself in balance, scientists believe. The various antibodies can either suppress or stimulate one another.
Because it has to be able to bind to the first antibody, this anti-antibody has a structure somewhat similar to the original antisense protein, which fits the self protein like a hand fits a glove. So the anti-antibody is also able to bind to the self protein, and it does so, triggering the autoimmune disease.
In 2001, Falk, Preston, and Pendergraft tested their theory of autoimmunity in mice. If it was happening as they thought, then when they injected a mouse with the antisense protein, the mouse should eventually make antibodies to the sense protein. That’s just what happened.
More evidence: from two patient blood samples, the researchers were able to isolate the two antibodies — one to the self protein, and one to the antisense protein. “We purified the antibodies and showed that they were separate and distinct entities,” Pendergraft says. They also did experiments to show that these two antibodies bound to each other.
The mystery is how, in human patients, the antisense protein gets inside the body. Is it produced there, or does it enter from outside? An intriguing possibility — the antisense protein is mimicked by a bacterium, fungus, or virus. The Carolina scientists found that several bacteria, including Staphylococcus aureus, a common culprit in food poisoning and one of the main pathogens linked to Wegener’s granulomatosis, carry pieces of protein similar to cPR3.
So it could happen like this: Staphylococcus or another bacterium invades. The patient’s body makes an antibody to kill the invader. Then the immune system produces a second antibody to clear the first, and that second antibody happens to bind to the self protein, triggering autoimmunity.
Not to say that every case of food poisoning will result in an autoimmune disease. Given that autoimmune diseases are relatively rare, other unknown factors must contribute.
“The theory is that genetic makeup and other factors come together,” Preston says, “and now you’ve set the stage that once you get this infection, everything’s poised to happen in this cascade.”
The researchers published their work in the January 2004 Nature Medicine. In an accompanying editorial, scientist Yehuda Shoenfeld of Israel’s Tel Aviv University calls the Carolina scientists’ study “elegant” and says that further study of their theory may improve treatment for a wide range of autoimmune disorders.
Pendergraft wrote his Ph.D. dissertation partly on this study, combining it with additional work he did related to PR3. These days, the team is exploring the theory further. They want to find out if patients with other autoimmune diseases harbor antibodies to antisense proteins. And they are searching blood samples from Wegener’s patients to identify the antisense protein that may have triggered their disease.
“We’re going to try to find it,” Preston says, “but we’re realistic enough to know that it may be long gone. It may have been there just transiently to start the cascade.” Falk says, “These findings give us a glimpse of how we can look for causes of autoimmune disorders, rather than just treating the consequences.”
Meanwhile, Dianne Shaw is taking a medicine called Imuran. She works full time, and she says that most days, thanks to her treatment, she feels fine. She says she gladly donated blood to be used in Falk’s studies. “The team’s discovery gives me great hope as a patient,” she says. “The possibility of more targeted therapy would change the lives of so many people suffering with this and other autoimmune diseases.”
Dianne Shaw is Director of Communications at Carolina’s Lineberger Comprehensive Cancer Center. She serves on the Board of Directors and as advocacy chair for the Wegener’s Granulomatosis Association. In March 2004 she spoke at a congressional briefing about the need for autoimmune-disease research.